
七岁男童罹患髓母细胞瘤,基因检测助力分子分型之余,检出MLH1胚系杂合变异确诊林奇综合征
2025-07-18 苏州绘真医学 苏州绘真医学 发表于上海

本病例是第三例林奇综合征合并髓母细胞瘤病例,为林奇综合征相关的癌症提供了额外证据,并为患者治疗开辟了新途径。
林奇综合征(Lynch syndrome,LS)是一种常染色体显性遗传病,其特征是DNA错配修复缺陷(dMMR),导致对多种癌症的易感性增加。林奇综合征患者的脑癌发病率范围为 2%-8%,其中胶质母细胞瘤、星形细胞瘤和少突胶质细胞瘤的风险最高。髓母细胞瘤(MB)是儿童常见的恶性脑肿瘤,而合并林奇综合征的髓母细胞瘤极为罕见。本文报告一例根据临床和病理结果诊断为髓母细胞瘤的儿童患者,通过下一代测序(NGS)和甲基化谱分析进一步确定为SHH活化、TP53突变型髓母细胞瘤。发现该患者肿瘤携带MSH2体细胞突变和疑似致病的MLH1胚系杂合变异。同时,肿瘤表现为高度微卫星不稳定(MSI-H)和异常升高的肿瘤突变负荷(TMB = 297.17 muts/Mb)。通过测序确认患者母亲和外祖母存在MLH1胚系变异,且患者外祖母有结直肠癌病史。最终,该患者被诊断为林奇综合征相关性髓母细胞瘤。本病例是第三例林奇综合征合并髓母细胞瘤病例,为林奇综合征相关的癌症提供了额外证据,并为患者治疗开辟了新途径。

▲摘自《2021年第五版WHO中枢神经系统肿瘤分类》
背 景
髓母细胞瘤(MB)是儿童最常见的恶性颅内肿瘤,占儿童颅内肿瘤的 25%。目前公认的髓母细胞瘤分子亚型主要包括四类:WNT亚型、SHH亚型、G3和G4。在回顾性队列中,胚系突变占髓母细胞瘤诊断的 6%。通常,结构性错配修复缺陷(CMMRD,constitutional mismatch repair deficiency,也译为体质性错配修复缺陷、先天性错配修复缺陷)比林奇综合征(LS)更常见于髓母细胞瘤。在CMMRD患者中,髓母细胞瘤约占中枢神经系统肿瘤的 10%。林奇综合征在髓母细胞瘤中极为罕见。
结构性错配修复缺陷(CMMRD)综合征是一种增加癌症易感性的遗传性疾病,由错配修复(MMR)基因(MLH1、PMS2、MSH2和MSH6)的两个等位基因均遗传有害突变引起。这些突变会损害DNA复制过程中MMR的正常功能,导致错配修复缺陷。CMMRD患者首次确诊癌症的中位年龄为 9.2 岁,其中中枢神经系统肿瘤是最常见的癌症类型。林奇综合征(LS)是一种常染色体显性遗传易感性疾病,其特征是DNA错配修复缺陷(dMMR),导致结直肠癌(CRC)、子宫内膜癌(EC)和其他癌症的发病风险增加。LS的确诊需要识别影响某个MMR基因(或EPCAM基因)的致病性或可能致病性胚系变异,目前LS这一术语仅限于符合该分子定义的病例。林奇综合征患者也可能发生脑肿瘤,在杂合MSH2(hMSH2)或hMLH1突变患者中的发生率为 0.88%,常见于 40 多岁人群。目前,仅有两例髓母细胞瘤病例携带遗传性杂合致病性MMR基因突变。
TIPS|CMMRD与林奇综合征
美国结直肠癌多学会工作组邀请专家制定了一份共识声明和建议,旨在帮助医护人员对双等位基因错配修复缺陷(BMMRD,biallelic mismatch repair deficiency)综合征(也称为结构性错配修复缺陷综合征[CMMRD,constitutional mismatch repair deficiency])患者进行适当的管理。
LS是一种常染色体显性遗传病,由DNA错配修复(MMR)或EPCAM基因的单等位基因胚系突变引起,是遗传性CRC的最常见原因。LS是由MLH1、MSH2、MSH6、PMS2和EPCAM中的大量杂合胚系突变引起的,肿瘤DNA的特点是微卫星不稳定性(微卫星高度不稳定 [MSI-H],或按惯例称为MSI)。LS相关癌症不完全外显;CRC的累积终生风险因突变基因和性别而异,对于MSH2、MLH1和MSH6基因,女性和男性的CRC累积风险分别为 40% 至 70%。与PMS2基因突变相关的LS患者CRC外显率显著降低,范围为 10% 至 20%。LS患者也易患结肠外恶性肿瘤,主要是子宫内膜癌(在携带MSH2和MLH1基因突变的女性中,发病率为40%),以及(在较小程度上)其他胃肠道和泌尿生殖系统肿瘤。这些综合征可以通过每年进行结肠镜检查和适当的妇科手术得到充分治疗。
双等位基因错配修复(MMR)基因突变(双等位基因错配修复缺陷 [BMMRD])会导致一种罕见且更致命的癌症综合征(OMIM数据库:2763000)。这种疾病也被称为结构性错配修复缺陷(CMMRD),因为出生时任何一个MMR基因的双等位基因失活的患者,其任何组织中均无DNA MMR活性。相比之下,在LS中,来自一个野生型等位基因的基因表达足以提供足够的DNA MMR活性,直到第二次突变使来自未受影响父母的野生型等位基因失活。由此产生的肿瘤组织存在DNA MMR缺陷,进而导致MSI的发生。

▲关于双等位基因错配修复缺陷(BMMRD)综合征的监测和管理建议:美国结直肠癌多协会工作组的共识声明[2]
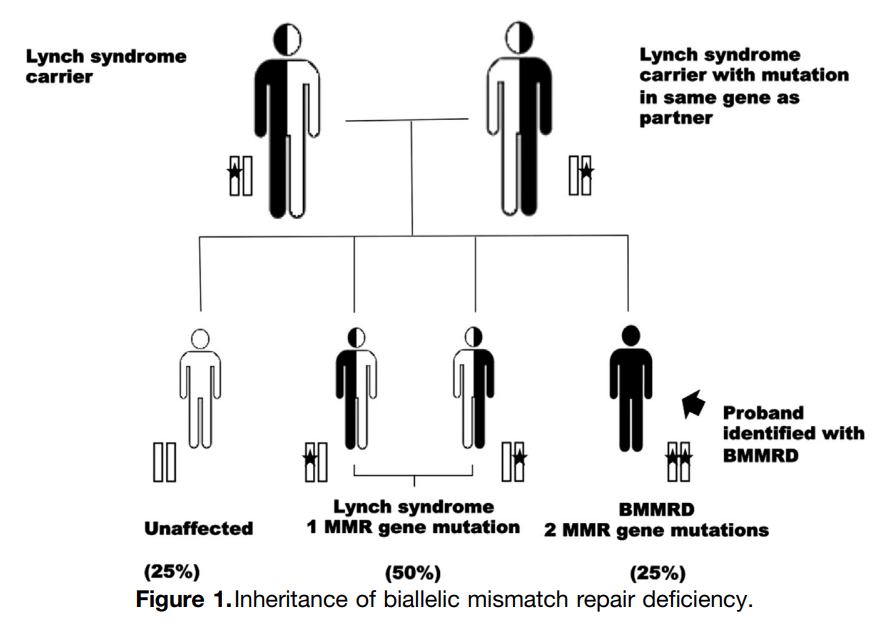
▲双等位基因错配修复缺陷(BMMRD/CMMRD)的遗传[3]
本文报告一名男孩患有髓母细胞瘤(MB),其肿瘤具有高度微卫星不稳定(MSI-H)、高肿瘤突变负荷(TMB),并通过NGS检测发现MSH2体细胞突变和MLH1胚系杂合突变。通过整合临床病理结果、NGS测序发现和甲基化分型结果,该患者最终被诊断为SHH活化、TP53突变型髓母细胞瘤。
病 例
患童男,7 岁,因间歇性头痛 2 周就诊于西安交通大学附属儿童医院。随后出现步态不稳和肢体震颤。既往无特殊病史或癌症病史。皮肤检查正常。询问患者家族史时,其外祖母在 54 岁时被诊断为结肠癌,其他家庭成员无癌症记录。头部CT扫描显示右侧小脑半球存在肿块。MRI检查显示右侧小脑半球有一个混杂信号肿块(6.5×6.0×3.6 cm),FLAIR成像显示信号强度轻度增高(图1A-D)。影像学检查提示诊断为髓母细胞瘤。随后,患者接受了手术治疗,实现了显微镜下完整切除,未见中枢神经系统转移迹象。术后,患童在手术部位接受了 54 Gy放疗,并接受了 36 Gy全中枢神经系统放疗,随后进行了 8 个周期的含铂三药化疗。经过 10 个月的治疗,目前患者已积极参与日常活动。
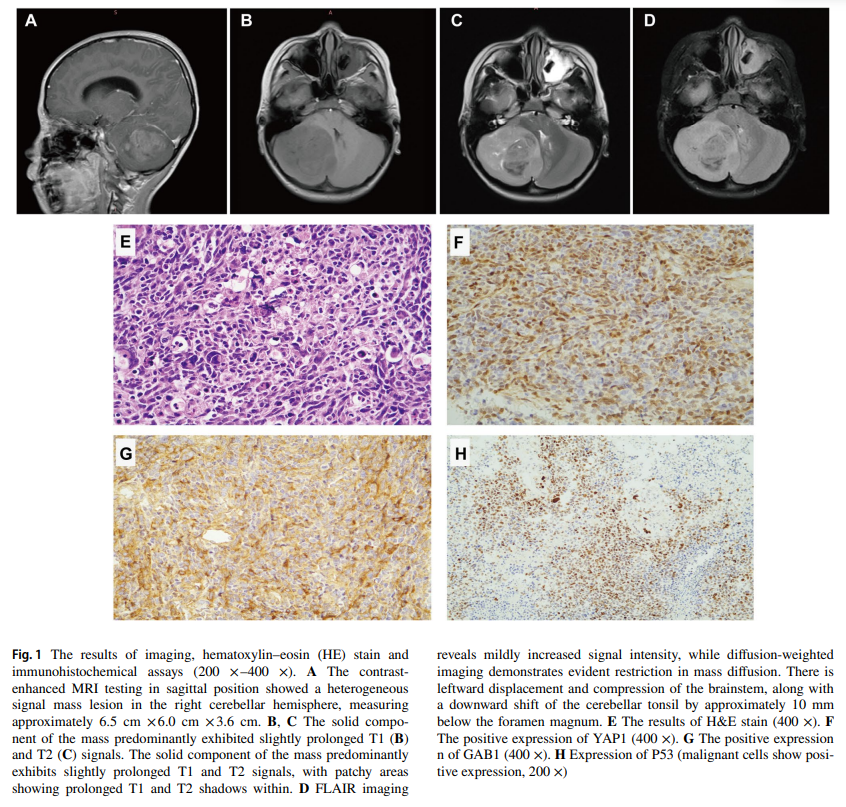
▲图1 影像学和病理检查结果
术后病理苏木精-伊红(HE)染色结果显示,肿瘤细胞弥漫分布,呈圆形、椭圆形或多边形。细胞核大且深染,伴有变性。细胞质极少或呈嗜酸性,伴有明显的间质血管增生,并可见核分裂象及假栅栏状坏死(图1E)。免疫组化(IHC)显示YAP1(+)、GAB1(+)、P53(恶性细胞阳性表达)(图1F-H)。此外,β-连环蛋白(细胞核-)、INI-1(+)、Ki-67(热点区域约 75%)。病理诊断为(后颅窝)小脑髓母细胞瘤(WHO IV级),组织学亚型符合大细胞/间变性髓母细胞瘤,分子亚型倾向于SHH活化型。随后对手术标本进行NGS和甲基化谱分析以明确分子亚型。结果显示患者存在 373 个体细胞突变,提示肿瘤突变负荷极高(TMB = 297.17 Mut/Mb)和MSI-H。MMR基因突变检测发现MSH2体系突变(c.1661+2 T>C,突变丰度 47.97%)及MLH1基因疑似胚系致病性杂合突变(p.E37Q,图2A)。MLH1 p.E37Q突变位于1号外显子,不在已知功能结构域内。该突变在人类基因组数据库中的频率极低。蛋白质功能预测软件Polyphen-2和SIFT的结果分别将其归类为“可能有害(D)”和“有害(D)”。此外,文献回顾已将该变异确定为可能致病性变异。林奇综合征(LS)的确诊需要识别影响某个错配修复(MMR)基因的致病性或可能致病性胚系变异。因此,基于上述分析及美国医学遗传学与基因组学学会(ACMG)威廉亚洲博彩公司 ,研究人员认为该MLH1胚系突变属于疑似致病性变异。通过血液样本对患者父母及外祖母进行了该MLH1胚系突变检测,结果显示其父未携带该突变(图2B),其母亲和外祖母均携带相同的MLH1(p.E37Q)杂合突变(图2C、D)。同时,患者存在体细胞TP53突变(c.538G>T,突变丰度 52.73%)和 5 个PTCH1突变(c.799G>T,突变丰度 13.35%;c.1576G>T,突变丰度 5.9%;c.2170G>T,突变丰度 5.59%;c.834G>A,突变丰度 4.76%;c.535缺失,突变丰度 3.73%),这些均为SHH型髓母细胞瘤中常见的突变。甲基化聚类结果与SHH-3型和SHH-4型均接近。拷贝数变异分析结果显示3p缺失和3q获得。结合免疫组化P53阳性表达及TP53突变的NGS测序结果,患者被综合判定为SHH-3型、TP53突变型髓母细胞瘤。SHH活化型髓母细胞瘤包括四个暂定分子亚组(SHH1、SHH-2、SHH-3、SHH-4),可通过DNA甲基化或转录组谱分析证实。SHH活化、TP53突变的髓母细胞瘤几乎均属于SHH-3型。该亚组患者往往年龄较小,以男性为主,组织学模式以大细胞变异型为特征。总体而言,该特定亚型与预后不良相关。
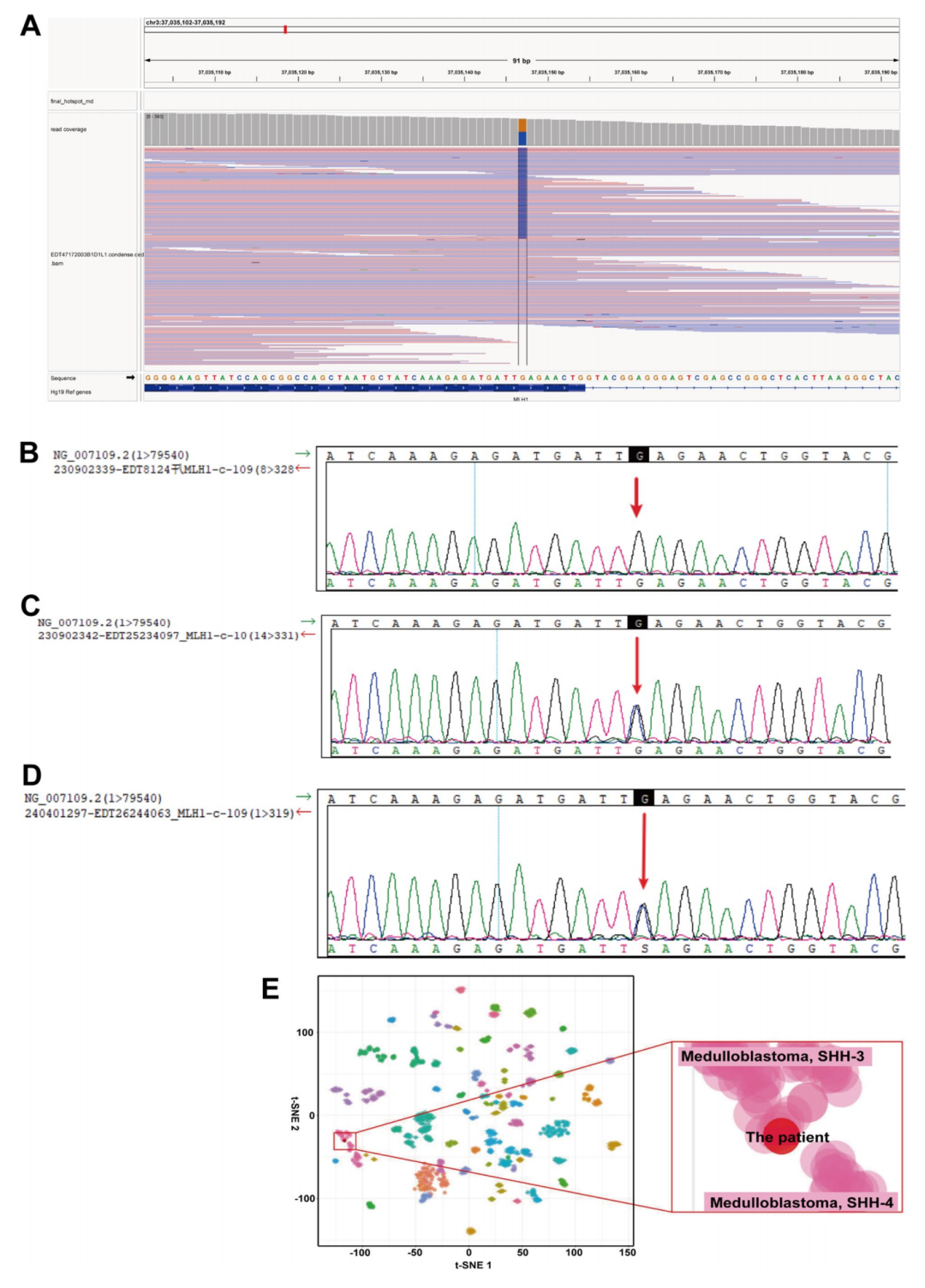

▲图2 基因检测和甲基化检测结果
讨 论
髓母细胞瘤(MB)是一组异质性的小脑胚胎性肿瘤,分为四个亚型。每个亚型均与独特的遗传改变、发病年龄和预后相关。本例初始临床病理诊断为SHH活化、TP53突变型MB。进一步甲基化谱分析显示患者为SHH-3型。精准分型后,患者的NGS检测结果提示微卫星高度不稳定(MSI-H)、MSH2体细胞突变及MLH1基因疑似胚系致病性杂合突变(p.E37Q)。基于临床和分子诊断,患者最终确诊为林奇综合征(LS)而非CMMRD。
与错配修复(MMR)基因胚系变异相关的表型表现取决于易感缺陷是杂合状态还是纯合状态。MMR基因的杂合变异是林奇综合征(LS)易感性的基础。MMR基因之一发生的双等位基因胚系突变可导致CMMRD,这是一种以常染色体隐性遗传方式遗传的儿童癌症易感综合征。在一项关于CMMRD的研究中,201 例CMMRD患者共报告 339 例癌症,其中中枢神经系统肿瘤最为常见(173 例[51%]);在这些中枢神经系统肿瘤中,髓母细胞瘤占 10%(18/173)。大多数报告CMMRD患者表现出多发性咖啡牛奶斑(CALMs),这些斑点边缘不规则,外观轻度弥漫;本例患者皮肤检查未发现皮肤疾病或异常。此外,CMMRD患者可能与跨代近亲结婚或近亲通婚相关,但本例家族无跨代近亲结婚或近亲通婚的记录。一项针对 773 例儿童实体瘤的研究显示LS患病率仅为 0.8%(6/773),且该研究未发现髓母细胞瘤与LS共存的病例。迄今为止,仅报告 2 例髓母细胞瘤合并LS的病例:其中一例来自病例系列报告,该报告描述了 5 例具有MMR基因胚系变异的患者,其中 4 例诊断为CMMRD,1 例(10 岁)诊断为LS(MSH2杂合突变)合并髓母细胞瘤;另一项病例报告中,一名男性髓母细胞瘤患者存在遗传性杂合致病性POLE变异和新发杂合致病性PMS2变异。错配修复缺陷(MMRD)和/或聚合酶校对基因(PP:POLE、POLD1)缺陷(PPD)可导致具有高单核苷酸变异或高肿瘤突变负荷(TMB)及微卫星不稳定(MSI)的癌症,这些共同构成DNA复制修复缺陷(RRD)的标志。因此,本例是合并PPD与LS的复杂RRD病例。
本文病例是第三例林奇综合征合并髓母细胞瘤的病例。对髓母细胞瘤基因组和甲基化谱的研究可能会修正遗传和肿瘤学诊断,并可能发现新的疾病实体;当结合胚系检测时,这些新实体需要细致的监测和个性化治疗方案。在本文病例中,研究人员通过临床病理诊断和下一代测序(NGS)检测,不仅准确确定了该髓母细胞瘤(MB)患者的分子亚型和甲基化特征,更关键的是明确了患者林奇综合征(LS)的诊断。这是第三例髓母细胞瘤合并林奇综合征的病例,拓宽了人们对林奇综合征相关肿瘤的认知。
我司可提供中枢神经系统肿瘤全基因组甲基化检测(基因芯片方法学),覆盖了CpG岛、启动子、编码区及增强子区域共约100万个CpG位点,筛选样本中的DNA甲基化位点,并与数据库中已知样本信息进行比较聚类分析,辅助中枢神经系统肿瘤的精准诊断及分子分型,同时包括了染色体拷贝数变异分析。
此外,还可增选我司髓母细胞瘤全外显子组基因检测,也可以送检髓母细胞瘤1299基因+染色体拷贝数变异检测、髓母细胞瘤690基因+染色体拷贝数变异检测或者脑肿瘤460基因+染色体拷贝数变异检测(NGS+CNV-Seq方法学),辅助临床诊断,预测可能获益的靶向、免疫及化疗药物,评估肿瘤遗传风险。
参考文献:
[1]Zheng H, Zhang G, Jiang B, Zhang L, Duan Q, Shi H. Medulloblastoma associated with Lynch syndrome: a case report of germline MLH1 variant and tumor molecular characterization. Invest New Drugs. 2025 May 19. doi: 10.1007/s10637-025-01527-6. Epub ahead of print. PMID: 40388014.
[2]Durno C, Boland CR, Cohen S, Dominitz JA, Giardiello FM, Johnson DA, Kaltenbach T, Levin TR, Lieberman D, Robertson DJ, Rex DK. Recommendations on Surveillance and Management of Biallelic Mismatch Repair Deficiency (BMMRD) Syndrome: A Consensus Statement by the US Multi-Society Task Force on Colorectal Cancer. Gastroenterology. 2017 May;152(6):1605-1614. doi: 10.1053/j.gastro.2017.02.011. Epub 2017 Mar 28. PMID: 28363489.
[3]Q Rana H, Syngal S. Biallelic Mismatch Repair Deficiency: Management and Prevention of a Devastating Manifestation of the Lynch Syndrome. Gastroenterology. 2017 May;152(6):1254-1257. doi: 10.1053/j.gastro.2017.03.013. Epub 2017 Mar 19. PMID: 28327367.

 分享
分享
本网站所有内容来源注明为“williamhill asia 医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于williamhill asia 医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“williamhill asia 医学”。其它来源的文章系转载文章,或“williamhill asia 号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与williamhill asia 联系,williamhill asia 将立即进行删除处理。
在此留言













#髓母细胞瘤# #林奇综合征#
2 举报